
Huntington’s disease (HD) is an autosomal dominant genetic disease that causes the neurons in the brain to degrade over time, which has a variety of effects (Datta 2024). I chose mostly to depict what the stages of the disease are and what it might look like as the disease progresses.
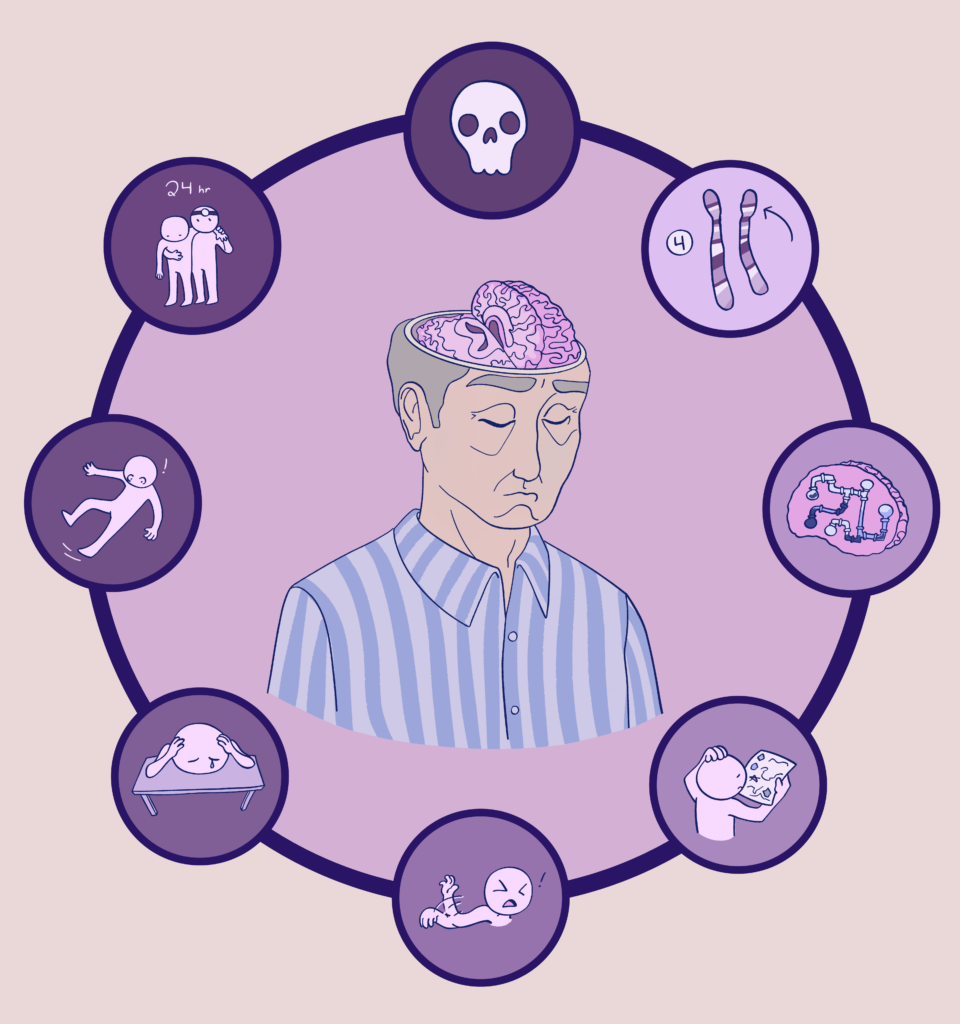
As this disease affects the nervous system and specifically the brain, the course objective I chose to relate this to was “Identify the various components and key structures of the nervous system” from the list provided in the STEAM projects module.
The earliest way to tell that someone has this disease is only possible through looking at their DNA. Huntington’s disease is characterized by excessive trinucleotide repeats on the short arm of the fourth chromosome, usually inherited from a parent (Datta 2024). Unaffected persons tend to have less than 35 CAG repeats, while affected persons have more than 36. The more repeats, the faster the onset of the disease and the quicker it progresses (Podvin et al. 2018). In my art I represent this by the chromosome pair labeled “4” at the 1 o’clock position.
This section of DNA codes for the Huntingtin protein, and its exact function is still unknown. In an unaffected brain, this protein has a relatively even distribution. The excess repeats seen in Huntington’s patients create a mutant misfolded form of this protein that is unable to fulfill its purpose and even begins to accumulate in areas of the brain, causing further damage (Datta 2024). I represent this idea by the next illustration in the clockwise direction which depicts a brain with clogged pipes in it.
This accumulation of proteins happens over time, and most patients only begin getting symptoms in their 30s or 40s, which get worse and worse as they get older. If the number of repeats is over 60, symptoms can appear before age 20 and it is called juvenile Huntington’s disease. Juvenile HD is less common, which is why I have made the patient in the middle of the circle an older person (Podvin et al. 2018). I cut open his brain mostly so that it is obvious that there is something wrong there.
HD affects the whole brain, but the most damage is seen in the areas of the brain that handle movement and cognition. The cognitive symptoms include impaired memory, difficulty organizing and executing tasks, loss of inhibition, and gain of behavioral changes (Datta 2024). I represent this with the next image clockwise featuring a person holding a failed attempt at a to-do list.
The movement symptoms most often result in chorea, which are involuntary muscle movements. This can make it difficult to walk, speak, and swallow (Mayo Clinic 2024). In the later stages this involuntary movement is replaced by uncontrollable rigidity of the muscles. The involuntary contractions interfere with purposeful movement, which becomes more and more difficult to execute (Datta 2024). I represent that with the next image clockwise showing a person alarmed at their arm movements, mostly to get across the point without words and not because motions like that are the most common.
Mental illness is often seen in HD patients, and is considered to be mostly because of neural decline and not just being faced with the prospect of a Huntington’s diagnosis. The most common one seen is depression, though OCD and mania have also been observed (Mayo Clinic 2024). I represent this in the next image clockwise with a person slumped sadly on their desk.
As the symptoms worsen, the person may fall frequently and their ability to care for themselves is diminished. They may also have seizures or tremors (Mayo Clinic 2024, Datta 2024). I represent this stage of the disease in the next image clockwise depicting a person mid fall.
In the end stages, weight loss is common and affected persons become unable to sustain a purposeful muscle contraction. This causes greater levels of disability, and they require round the clock care (Datta 2024, Podvin et al. 2018). I represent this in the next image with a patient being supported by someone wearing a doctor’s head mirror. Above them I wrote 24hr to indicate the constant nature of the required care.
Death usually occurs around 15-20 years after the symptoms begin, once the damage to the brain is severe enough to no longer support life (Podvin et al. 2018). I represent this with the skull, a common symbol of death.
This disease is incurable and medications are only used to alleviate the symptoms, which is why I did not include them in my drawing (Datta 2024).
Sources:
Datta, N. (2024). Molecular mechanisms and clinical features of Huntington Disease: A fatal neurodegenerative disorder with autosomal dominant inheritance. Columbia Undergraduate Science Journal, 17(1), 17–34. https://doi.org/10.52214/cusj.v17i1.10288
Podvin, S., Reardon, H. T., Yin, K., Mosier, C., & Hook, V. (2018). Multiple clinical features of Huntington’s disease correlate with mutant HTT gene CAG repeat lengths and neurodegeneration. Journal of Neurology, 266(3), 551–564. https://doi.org/10.1007/s00415-018-8940-6
Mayo Foundation for Medical Education and Research. (2024, April 25). Huntington’s disease. Mayo Clinic. https://www.mayoclinic.org/diseases-conditions/huntingtons-disease/symptoms-causes/syc-20356117
Moey’s STEAM project showcases the stages of Huntington’s disease through various illustrations. Huntington’s disease affects the brain by causing excess repeats of the Huntingtin protein and an uneven protein distribution. This disease affects mainly the areas of the brain that are responsible for movement and cognition. The illustration in the center of the image shows an older gentleman because Huntington’s disease is more common in older people and symptoms worsen with age. As for the illustrations surrounding the centerpiece, they start with an image of chromosomes which symbolize Huntington’s disease affecting the fourth chromosome. The brain with pipes illustrates the Huntingtin protein increasing in areas of the brain, creating an uneven protein distribution. The next illustrations are symptoms that occur and can be noticed over time with the disease. The character is confused when looking at a to-do list, which represents impaired memory, organization, and completing tasks, as well as behavioral changes. Next, involuntary muscle movements are common in the earlier stages which then become uncontrollable muscle movements. Depression is illustrated in the fifth bubble because mental illness is a common symptom in HD patients. Additionally, as symptoms worsen, falling frequently occurs and the ability to care for oneself decreases, further requiring around-the-clock care. The last image, a skull, illustrates that once the symptoms begin and the brain becomes too severely damaged, death is common around 15-20 years after symptoms begin.