
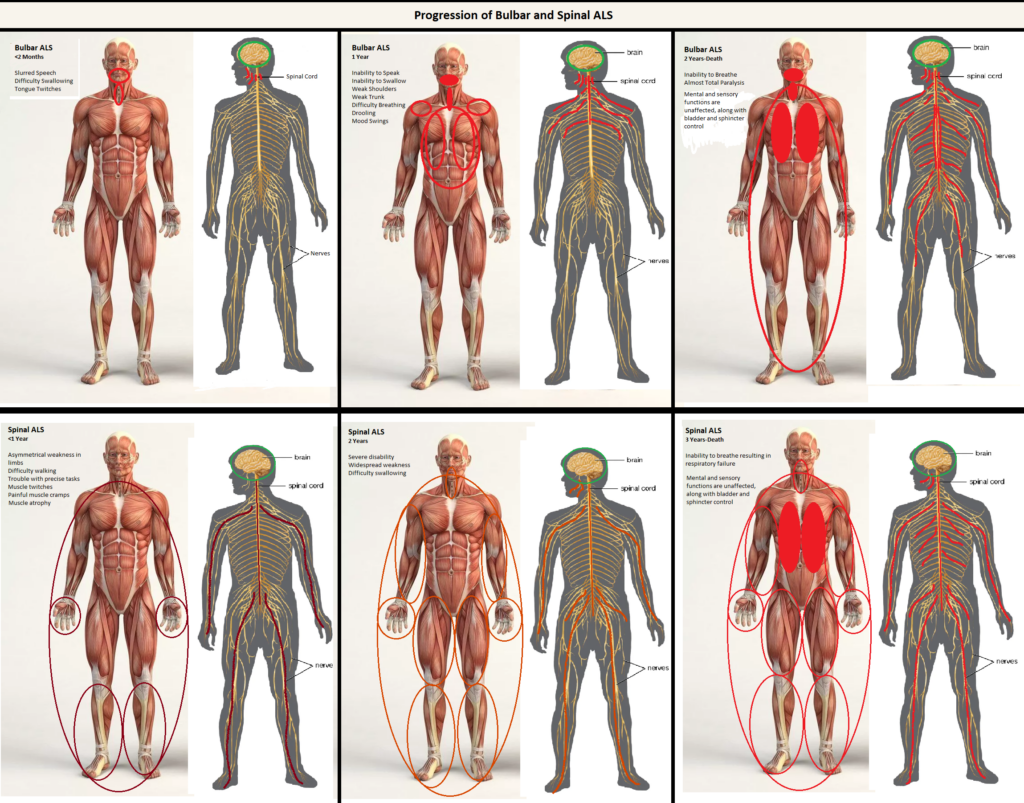
My project targets the learning objective that involves describing how a muscle contraction is induced, and I have chosen to cover amyotrophic lateral sclerosis (ALS) to depict this objective. ALS is a progressive degenerative condition that targets the anterior horn cells in the spinal cord and causes paralysis. It is currently uncurable and mostly untreatable. My artwork depicts the progression of both the spinal and bulbar forms of ALS.
ALS causes paralysis by degrading the anterior horn cells on the spinal cord, which is the origin point for motor neurons that innervate skeletal muscle. This causes an inability to properly contract skeletal muscles due to the lack of a functioning motor neuron to carry the action potential to the axons at the muscles. ALS does not affect sphincter or bladder control, brain function, or sensory function, as these are not related to the anterior horn cells or skeletal muscle.
The first row of my project shows how the bulbar form of ALS progresses throughout the body by showing where the anterior horn cells on the spinal cord start to deteriorate, and what they affect by doing so. In the first block of the art is the illustration of the symptoms related to the first 2 months of the bulbar form of ALS. In this period of time, deterioration has only just begun, but is already causing very noticeable issues. The bulbar form of ALS tends to start at the head and work its way down the body, so the first symptoms can be difficulty speaking, slurred words, difficulty swallowing, and drooling as a result of the difficulty swallowing combined with excessive salivation. The deterioration of anterior horn cells in the upper regions of the spine cause these effects, but as the condition progresses down the spine, the symptoms become more serious. The second block in my art covers the symptoms up to the 1-year mark. This includes many upper body issues, such as shoulder weakness, upper trunk weakness, an inability to swallow and speak, difficulty breathing, and rapid mood swings. By the two-year mark, most patients with the bulbar form of ALS are dead due to an inability to breathe and almost total paralysis. Throughout the entire progression of the condition, the patient will retain all mental functionality, sphincter and bladder control, as well as sensory reception.
The second row of the art illustrates the spinal form of ALS, which is a much slower progressing version that first manifests symptoms in the extremities. This is due to a lower spinal deterioration of the anterior horn cells that allow the extremities to be innervated. This is depicted in my project and different colors are used at the different stages to show the slower progression of this form of ALS. In the first year a patient will experience asymmetrical muscle weakness, resulting in progressive muscle atrophy. They will also experience painful muscle cramps and have difficulty walking, along with trouble executing tasks that require dexterity with the hands, such as writing, by the end of the first year. In the second year, which is shown in the second block of my art, more widespread disability starts to take hold. Muscles have atrophied severely and are almost paralyzed, but still somewhat functional. Difficulty swallowing and breathing means that most patients require a gastrostomy tube for nutrition. Many patients will die within the third year, when complete inability to breathe causes respiratory failure. The same functions are preserved as in the bulbar form.
Sources:
- Walling, A. D. (1999, March 15). Amyotrophic lateral sclerosis: Lou Gehrig’s disease. American Family Physician. Retrieved November 22, 2022, from https://www.aafp.org/pubs/afp/issues/1999/0315/p1489.html#afp19990315p1489-b8
- Calvo, A. C., Manzano, R., Mendonça, D. M. F., Muñoz, M. J., Zaragoza, P., & Osta, R. (2014, August 3). Amyotrophic lateral sclerosis: A focus on disease progression. BioMed Research International. Retrieved November 22, 2022, from https://www.hindawi.com/journals/bmri/2014/925101/
- Boillee, S. (2006, June 2). Onset and progression in inherited ALS determined by motor … – science. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Retrieved November 22, 2022, from https://www.science.org/doi/10.1126/science.1123511
- Centers for Disease Control and Prevention. (2022, May 13). CDC – amyotrophic lateral sclerosis: About. Centers for Disease Control and Prevention. Retrieved November 22, 2022, from https://www.cdc.gov/als/WhatisAmyotrophiclateralsclerosis.html
The unit objective used for this project is to describe how a muscle contraction is induced. I feel that you did a good job explaining how ALS affects muscle contraction early on in your writing. The choice to explain how ALS affects muscle contractions early on gave you the opportunity to you to explain further using your images as a reference. I really enjoyed that you used your images as a reference point. It was nice to be able to look at exactly what you were talking about. I also like how each stage of the disease has different areas affected circles or outlined in your art piece. I think you did a good job of explaining in depth the different stages of deterioration of the anterior horn cells of the upper spinal cord through the progression of Bulbar ALS and the deterioration of the lower anterior horn cells and how they affect the extremities in someone with Spinal ALS. I also think you did a good job of getting all of the symptoms associated with each progression of the disease in both spinal ALS and Bulbar ALS. I found this really interesting because I did not know much about ALS before reading this, especially not that there was more than one kind of ALS. I also find it intriguing that the progression of this disease is so fast in bulbar ALS. Following the fast pace of the progression of bulbar ALS, I found it intriguing that the progression of Spinal ALS is significantly slower. I feel as if you put in a lot of time and effort into this, very well done in my eyes.