
For this project, I will be focusing on Huntington’s disease and its relation to the course objective “Identify the various components and key structures of the nervous system” from unit six. It will concentrate on Huntington’s disease and its effect on nervous system structures, specifically regions of the brain.
Individuals generally experience the onset of Huntington’s disease in their 30s or 40s, but the age of onset can vary greatly. Huntington’s disease is a rare genetic disorder that impacts more than 15,000 Americans currently with the disease, and many more are at risk of experiencing it (Johns Hopkins Medicine, n.d.). In the early stages of the disease, individuals may experience mood swings, depression, personality changes, impaired judgment, and forgetfulness. However, as the disease processes, symptoms worsen and affect the individual’s ability to walk, talk, and swallow. A person with Huntington’s disease also experiences uncontrolled movements known as chorea (Huntington’s Disease Society of America, n.d.).
Huntington’s disease is a brain disorder that causes brain cells and neurons in specific regions of the brain to break down and die (Johns Hopkins Medicine, n.d.). This disease is caused by a genetic mutation in the huntingtin gene (HTT) located on chromosome 4. The mutation leads to the huntingtin protein containing an excessively long polyglutamine repeat. As a result, CAG (genetic code for glutamine) will recur repeatedly in the DNA sequence, which puts an individual at risk of developing the disease (McColgan & Tabrizi, 2017). Those with 36 to 39 repeats have a chance of getting the disease, but those with over 39 CAG in their DNA are at a significant risk of developing Huntington’s disease. Consequently, this mutation causes the proteins to grow abnormally in brain cells, which results in them becoming damaged and dying (Huntington’s Disease Society of America, n.d.).
The neurodegeneration of Huntington’s disease primarily affects the central nervous system (CNS), especially the brain. The area of the brain most affected by Huntington’s disease is the basal ganglia, which consists of the striatum, globus pallidus, and subthalamic nucleus. The basal ganglia regulates movement, motor control, and emotional responses. The degeneration of neurons in the structures of the basal ganglia causes them to shrink, which interferes with their normal function. This results in the individual experiencing difficulty moving and having uncontrollable movements (Reiner, Dragatsis, & Dietrich, 2011). Huntington’s disease also impacts the cerebral cortex, cerebellum, frontal lobe, and thalamus. All of these structures have important functions. The cerebral cortex is involved in higher thought processes like memory, thinking, language, and emotions, while the cerebellum aids coordination and balance. The frontal lobe is responsible for language, emotion, and decision-making, whereas the thalamus is essential for relaying information. As a result of the loss of brain cells and neurons in Huntington’s disease, these brain structures undergo shrinkage, causing disruptions in their functions (Reiner, Dragatsis, & Dietrich, 2011).
At this time, there is no cure for Huntington’s disease, but there are treatments to help manage and slow the progression of the disease. For individuals who experience motor issues like chorea, medications such as tetrabenazine, deutetrabenazine, tiapride, and risperidon aid in reducing involuntary movements (Eileeng et al., 2022). Those with Huntington’s disease also commonly have signs of mood swings, anxiety, and depression, which are treated with antidepressants and antianxiety medications (Johns Hopkins Medicine, n.d.). It is recommended to do physiotherapy, speech therapy, and psychotherapy, in addition to taking medication to improve mobility, communication, and emotional health. Experimental treatments are also being studied. One experimental treatment is antisense oligonucleotides (ASOs), which aid in blocking the production of the harmful Huntington protein (Eileeng et al., 2022).
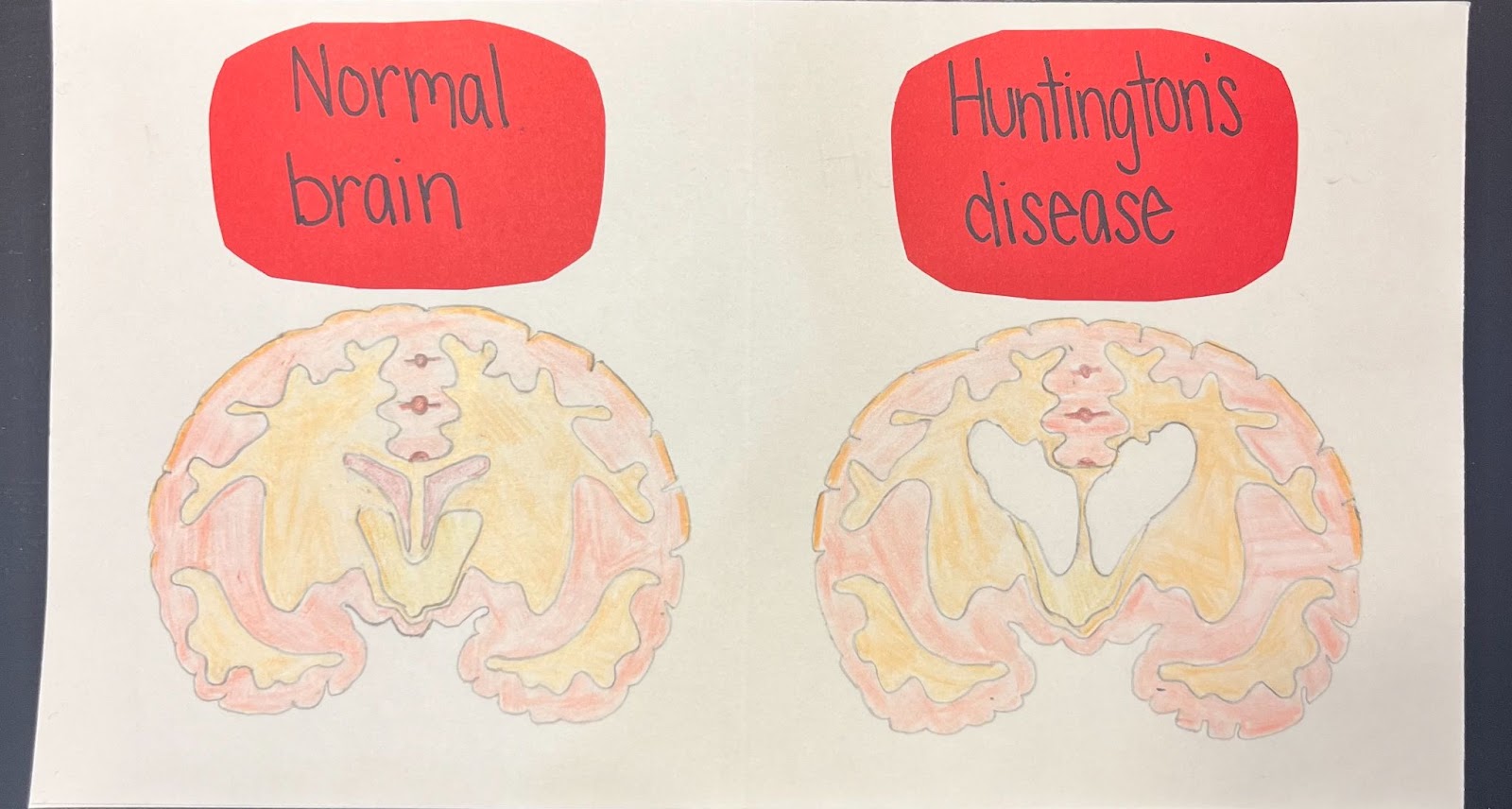
My artwork displays a visual comparison between a normal brain and a brain with Huntington’s disease. On the left, it shows a healthy brain, which has no signs of degeneration or shrinkage. On the right is a brain with Huntington’s disease. It reflects how the loss of brain cells and neurons has resulted in regions of the brain shrinking. This portrays the problems of motor, emotional, and cognitive decline developed as a result of Huntington’s disease.
References
Eileeng, M., Camila, H., Ledys, M., Karin, M., María, M., Natalia, D. V., Xilene, M., & Alexander, R. (2022). Current knowledge and future directions in Huntington’s disease. Archivos de Neurociencias, 27(4), 31–43. https://doi.org/10.31157/an.v27i4.346
Healthdirect Australia. (n.d.). Huntington’s disease. Healthdirect Australia. Retrieved November 17, 2024, from https://www.healthdirect.gov.au/huntingtons-disease#caused
Huntington’s Disease Society of America. (n.d.). Overview of Huntington’s disease. Huntington’s Disease Society of America. Retrieved November 17, 2024, from https://hdsa.org/what-is-hd/overview-of-huntingtons-disease/
Johns Hopkins Medicine. (n.d.). Huntington’s disease. Johns Hopkins Medicine. Retrieved November 17, 2024, from https://www.hopkinsmedicine.org/health/conditions-and-diseases/huntingtons-disease#:~:text=Huntington%20disease%20is%20a%20brain,intellectual%20abilities%2C%20and%20uncontrolled%20movements.
McColgan, P., & Tabrizi, S. J. (2017). Huntington’s disease: A clinical review. European Journal of Neurology, 24(9), 1289-1297. https://doi.org/10.1111/ene.13413
Reiner, A., Dragatsis, I., & Dietrich, P. (2011). Genetics and neuropathology of Huntington’s disease. International review of neurobiology, 98, 325–372. https://doi.org/10.1016/B978-0-12-381328-2.00014-6
Hailey’s Steam project is on Huntington’s disease, It ties into Chapter six of our anatomy class which covers the basic design of the nervous system, her project focuses on how the disease effects the brain specifically.
In a nutshell, HD is hereditary genetic mutation, which starts by attacking the Basal Ganglia and cause severe degradation of both fine and gross motor skills along with emotional regulation. Unfortunately the disease process moves onto other major structure in the brain and will eventually interfere with cognitive function. Hailey mentioned that people typically see symptoms as early as in their 30s. I have a friend who’s mother was diagnosed with HD but is only in early stages, he mentioned that if you have the genetic predisposition for it like he does that they will only allow you to be tested for if you meet with a counselor first.
Thankfully Hailey wrote a little hope into her project, bring up some of the options that are being researched to at least slow the effects of HD.
Hailey was very thorough and did some excellent research for her project, it is well written, informative, and cited.
Hailey displayed her creative talents through art and illustrated the jarring difference HD makes in one’s brain.
A+ from me for a well done project.