
Class Objective: Compare and contrast the organelles and their functions
Lysosomes are an interesting organelle that are membrane enclosed and essentially function as the garbage disposal system of the cell. They act to break down and process all kinds of things within the cell including bacteria, viruses and toxins. To do this, they are full of digestive enzymes that can help degrade these things along with other organelles that may be damaged. The interior of lysosome is, therefore, known to be caustic and can damage other areas of the cell if released. Lysosomes have several other functions of no less importance, one of which is the breakdown and release of glycogen.
Glycogen is synthesized in the muscle and liver cells, and it is an easily available form of glucose that can provide energy rapidly to the cells. In basic terms, glycogen helps to keep our muscles fueled during exercise. There are several mechanisms that trigger the breakdown of glycogen inside of our bodies, and one of them is the release of adrenaline through the “fight or flight” response. (Patino & Orrick., 2024) Glycogen storage in skeletal muscle serves an especially critical function in terms of rapid ATP (adenosine triphosphate) generation, and there is a very close relationship between muscle resistance to fatigue and these levels. (Patino & Orrick., 2024) Additionally, it is known that glycogen metabolism is extremely important during fasting as well as muscle contraction. (Adeva-Andany, González-Lucán, Donapetry-García, Fernández-Fernández, & Ameneiros-Rodríguez, 2016) It is imperative for human cellular energy demands that glucose homeostasis is maintained via the glycogen conversion mechanism. (Kanungo, Wells, Tribett, & El-Gharbawy, 2018)
When this critical node in cellular energy production is interrupted, the human body has problems, and the results can be fatal. Pompe (pom-pay) Disease, or GSD2 (glycogen storage disorder #2), is the common term for an autosomal recessive genetic disorder that results in the accumulation of glycogen within the lysosomes themselves. Mutations in the GAA gene locus is what causes this disorder and although the mechanism of interruption is not completely understood, it is believed that acid maltase is the chemical which accomplishes the breakdown. (Adeva-Andany, González-Lucán, Donapetry-García, Fernández-Fernández, & Ameneiros-Rodríguez, 2016) When glycogen begins to build up within the lysosomes, this leads to a whole cascading set of issues for the cells involved and, thus, the human body. The buildup of glycogen means that the body has less glycogen to convert into glucose for energy. The increasing levels of glycogen within the lysosomes can and will eventually lead to a rupture of the lysosome. (Labella, et al., 2023) This rupture releases all kinds of caustic material into the cell that can lead to tissue damage and the death of the cell. The resultant eruption is a mixed cellular magma of damaged organelles, decaying viruses, dead bacteria, and acidic digestive juices. Furthermore, in one study it was found that lysosomal glycogen accumulation promotes more glycogen synthesis which results in further aggravation of this disease. (Canibano-Fraile, et al., 2022) This cycle of buildup and eruption leads to a complex pathogenesis that is not simply related to the damage to the lysosomes, but also implies a cascade of other complex issues that involve things such as regulation of signaling pathways, oxidative stress, and mitochondrial abnormalities, among other things. (Labella, et al., 2023)
There are two forms of this disease, and both have serious implications. The main difference between the two forms is when the onset of symptoms is observed. The infantile version of Pompe Disease usually occurs in infancy and certainly before one year of age. Symptoms include progressive weakness of the muscles and possibly enlargement of the heart, liver or tongue. It is also expected that difficulty in breathing would be observed. In late onset Pompe Disease, there is a steady weakening of the legs and trunk as well as muscle pain over large areas. Patients may lose the ability to exercise, and they may fall frequently. Also included in the typical list of symptoms is shortness of breath, and/or frequent respiratory infections. The prognosis for patients with infantile Pompe Disease is usually not good, although enzyme replacement therapy (ERT) has extended survival rates for some patients. Late onset Pompe Disease in adults is still a multi-system disease that requires a multidisciplinary approach to management, however it is much more survivable. (Labella, et al., 2023)
Interestingly, in the past ten years muscle MRI techniques have significantly developed to the point that this can be an important tool for diagnoses and maintenance when looking at overall skeletal muscle fat fractions. (Labella, et al., 2023) This technique of diagnosis can be especially useful for those who suffer from late onset Pompe disease, and when combined with ERT, this course of treatment often leads to desirable outcomes. Additionally, gene therapy holds great promise for the future treatment of both infantile and late onset Pompe Disease and may help to stem the tide of this potentially fatal eruption in lysosomes.
Works Cited
Adeva-Andany, M. M., González-Lucán, M., Donapetry-García, C., Fernández-Fernández, C., & Ameneiros-Rodríguez, a. E. (2016, June 5). Glycogen metabolism in humans. Retrieved from BBA Clinical: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4802397/
Canibano-Fraile, R., Harlaa, L., Santos, C. A., Hoogeveen-Westerveld, M., Demmers, J. A., Snijders, T., . . . van, A. T. (2022, October 17). Lysosomal glycogen accumulation in Pompe disease results in disturbed cytoplasmic glycogen metabolism. Retrieved from Journal of inherited metabolic disease: https://pubmed.ncbi.nlm.nih.gov/36111639/
Kanungo, S., Wells, K., Tribett, T., & El-Gharbawy, a. A. (2018, December). Glycogen metabolism and glycogen storage disorders. Retrieved from Annals of translational medicine: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6331362/
Labella, B., Piccinelli, S. C., Risi, B., Caria, F., Damioli, S., Bertella, E., . . . Filosto, a. M. (2023, September). A Comprehensive Update on Late-Onset Pompe Disease. Retrieved from Biomolecules: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10526932/
Patino, S. C., & Orrick., J. A. (2024, January 27). Biochemistry, Glycogenesis. Retrieved from StatPearls [Internet]: https://www.ncbi.nlm.nih.gov/books/NBK549820/
*Art: The associated art piece below is a 2-part watercolor study meant to evoke imagery we normally associate with volcanoes. The combination of similar word associations and suggestive imagery in the essay, when combined with the art, should give the reader a lasting understanding of Pompe Disease by associating it with the famous Pompeii eruption.
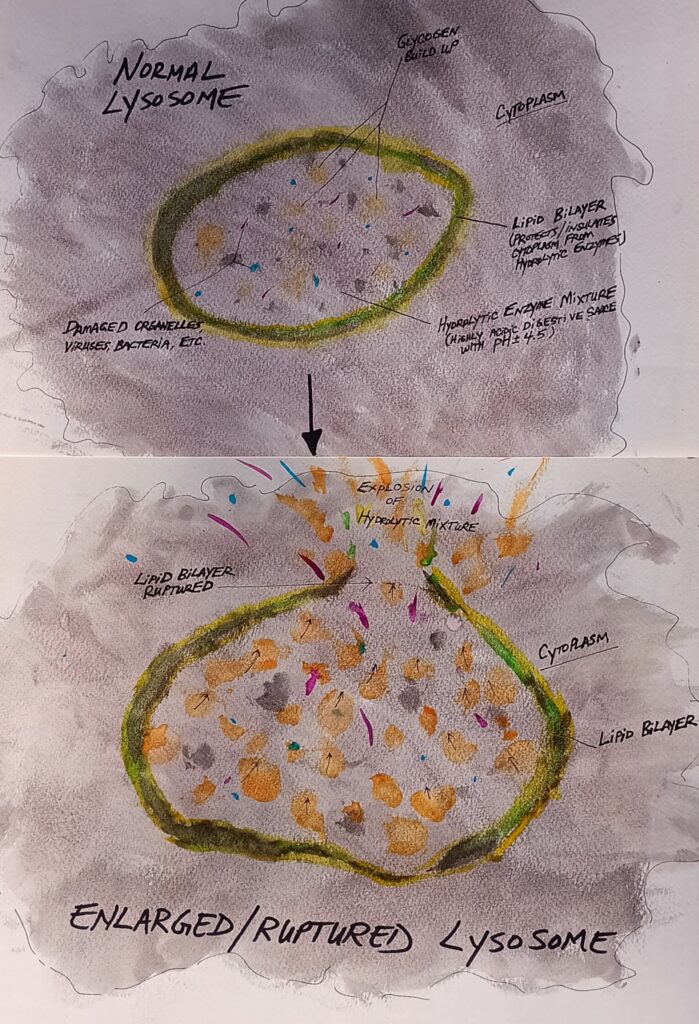
Matt steam project was on the lysosome and the effect it has on pompe disease. Lysosomes are an organelle that works as a garbage disposal. They break down and process cells. One of the cells they break down is glycogen. Glycogen, is a glucose synthesized in muscle and liver cells, provides energy for muscle during exercise, aiding in rapid ATP generation and muscle resistance to fatigue. Pompe Disease is an autosomal recessive genetic disorder causing the accumulation of glycogen within lysosomes. The accumulation of glycogen in lysosomes can lead to cell rupture, releasing caustic material, tissue damage, and cell death. This accumulation also promotes glycogen synthesis, further aggravating the disease, as it reduces the body’s ability to convert glycogen into glucose. There are two different types of pompe disease, the infantile which occurs in the first year of infancy causing muscle weakness, heart, liver, or tongue enlargement, and difficulty breathing. The other type is late onset pompe disease causing weakness of legs and muscle pain in larges areas. In Matt’s watercolor interpretation of normal lysosome and ruptured lysosomes I found he did a very good job showing what is inside a normal lysosome such as the glycogen and how normal glycogen is supposed to build up. In the rupturing lysosome I could visionally see the lipid bilayer breaking and having the glycogen coming out of the lysosome gave me better vision understanding on how the lysosome would look if the event of a rupture. Matt’s steam project was very in depth and very easy to understand going through the function of the lysosome and how it related to pompe disease. Amazing job Matt!